司 法 鉴 定 技 术 规 范
SF/Z JD0107022——2018
毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的液相色谱-串联质谱检验方法
Determination of Δ9-tetrahydrocannabinol, cannabidiol and cannabinol in hair by liquid chromatography-tandem mass spectrometry
2018-11-08 发布 2019-01-01 实施
中华人民共和国司法部公共法律服务管理局 发 布
目 次
前言
1. 范围
2. 规范性引用文件
3. 术语和定义
4. 原理
5. 试剂、仪器和材料
6. 操作方法
7. 分析结果评价
附录 A(资料性附录) 毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的 MRM 色谱图
附录 B(资料性附录) 方法学有效性验证数据
表 1 流动相梯度洗脱程序
表 2 Δ9-四氢大麻酚、大麻二酚、大麻酚和内标物的定性离子对、定量离子对和保留时间
表 3 相对离子对丰度比的最大允许相对误差
前 言
本技术规范按照GB/T 1.1-2009给出的规则起草。
本技术规范由司法鉴定科学研究院提出。
本技术规范由司法部公共法律服务管理局归口。
本技术规范起草单位:司法鉴定科学研究院。
本技术规范主要起草人:向平、沈敏、刘伟、沈保华、卓先义、严慧、吴何坚。
本技术规范的附录 A、B 为资料性附录。
本技术规范为首次发布。
毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的液相色谱-串联质谱检验方法
1 范围
本技术规范规定了毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的液相色谱-串联质谱(LC-MS/MS)
检验方法。
本技术规范适用于毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的液相色谱-串联质谱定性与定量分析。
2 规范性引用文件
下列文件对于本文件的应用是必不可少的。凡是注日期的引用文件,仅注日期的版本适用于本文件。
凡是不注日期的引用文件,其最新版本(包括所有的修改单)适用于本文件。
GA/T 122 毒物分析名词术语
GB/T 6682 分析实验室用水规格和试验方法
3 术语和定义
GA/T 122 中界定的术语和定义适用于本技术规范。
4 原理
毛发样品经清洗、冷冻研磨后,以甲醇超声法提取,用液相色谱-串联质谱的多反应监测(MRM)模式检测。经与平行操作的空白样品和添加样品对照,以保留时间、质谱特征碎片离子峰和离子对相对丰
度比进行定性分析;以峰面积为依据,采用内标法进行定量分析。
5 试剂、仪器和材料
5.1 试剂
本技术规范试验用水为一级水(见GB/T 6682规定),所用试剂:
- a) 甲醇:HPLC级;
- b) 乙腈:HPLC级;
- c) 50%甲酸溶液:HPLC级;
- d) 乙酸铵:HPLC级;
- e) 丙酮:分析纯;
- f) 20mmol/L乙酸铵溶液(含 1%甲酸):分别称取 1.54g 乙酸铵和 2mL 甲酸溶液置于 1000mL
容量瓶中,加水定容至刻度,pH 值约为 4;
- g) 内标为甲氧那明或其它合适内标物;
- h) 标准物质溶液:
1) 100µg/mLD9-四氢大麻酚、大麻二酚、大麻酚标准溶液:市售D9-四氢大麻酚、大麻二酚、
大麻酚标准物质溶液均置于冰箱中冷冻保存,保存时间 12 个月;
2) Δ9-四氢大麻酚、大麻二酚、大麻酚标准工作液:试验中所用其他浓度的标准工作溶液均
由 100µg/mL Δ9-四氢大麻酚、大麻二酚、大麻酚标准溶液用甲醇稀释而得,置于冰箱中冷
藏保存,保存时间 6 个月;
- i) 内标甲氧那明标准溶液:
1) 1.0mg/mL 甲氧那明标准储备溶液:精密称取甲氧那明 10mg 于 10mL 容量瓶中,加入适
量甲醇溶解并定容至刻度,配制成 1.0mg/mL 甲氧那明标准储备溶液。密封,置于冰箱中
冷冻保存,保存时间 12 个月;
2) 1ng/mL 甲氧那明标准工作溶液:移取 1.0mg/mL 甲氧那明标准储备溶液适量至容量瓶中,
加入甲醇稀释,混匀,配制成 1ng/mL 的甲氧那明标准工作溶液。密封,置于冰箱中冷藏
保存,保存时间 3 个月。
5.2 仪器和材料
仪器和材料包括:
- a) 液相色谱-串联质谱仪:配有电喷雾离子源(ESI);
- b) 电子分析天平:感量1mg;
- c) 离心机;
- d) 超声波清洗仪;
- e) 恒温水浴锅;
- f) 冷冻研磨仪;
- g) 移液器。
6 操作方法
6.1 定性分析
6.1.1 样品前处理
6.1.1.1 案件样品
毛发样品依次用适量的水和丙酮振荡洗涤两次,晾干后剪成约1mm段,置冷冻研磨仪中磨碎,呈
粉末状。
称取毛发粉末样品20mg,加入1.0mL内标甲氧那明标准工作液(甲氧那明1ng/mL),超声30min,
离心,转移上清液,于60℃水浴空气流下吹干。残留物用100mL甲醇复溶,供仪器检测。
6.1.1.2 控制样品
称取空白毛发粉末样品20mg两份,一份作为空白样品,一份添加Δ9-四氢大麻酚、大麻二酚、大麻
酚标准工作溶液,制得0.05ng/mg毛发添加样品,余下同6.1.1.1,与案件样品平行操作。
6.1.2 仪器检测
6.1.2.1 仪器条件
6.1.2.1.1 液相色谱条件
以下为参考条件,可根据不同仪器实际情况进行调整:
- a) 色谱柱:RestekAllure® PFP Propyl 五氟苯基柱(或其它等效柱),100mm ´1mm ,5mm
注:Restek Allure® PFP Propyl柱为美国Restek公司产品的商品名称,给出这一信息是为了方便本技术规范的使用者,
并不是表示对该产品的认可。如果其他等效产品具有相同的效果,则可使用这些等效产品。
- b) 流动相:A为 20mmol/L 乙酸铵和 1 %甲酸缓冲液,B 为乙腈,流动相梯度洗脱程序见表 1;
表1 流动相梯度洗脱程序
|
时间(min)
|
流动相A(%)
|
流动相B(%)
|
|
0
|
50
|
50
|
|
2.0
|
5
|
95
|
|
5.0
|
5
|
95
|
|
5.1
|
50
|
50
|
|
10.0
|
50
|
50
|
- c) 流速:300mL/min;
- d) 柱温:室温;
- e) 进样量:5µL。
6.1.2.1.2 质谱条件
以下条件作为参考,可根据不同仪器实际情况进行调整:
- a) 离子源:电喷雾电离-正离子模式(ESI+);
- b) 检测方式:多反应监测(MRM);
- c) 离子源电压(IS):5500V;
- d) 碰撞气(CAD)、气帘气(CUR)、雾化气(GS1)、辅助气(GS2)均为高纯氮气,使用前调节各气流
流量以使质谱灵敏度达到检测要求;
- e) 去簇电压(DP)、碰撞能量(CE)等电压值应优化至最佳灵敏度。
在以上色谱、质谱条件下,Δ9-四氢大麻酚、大麻二酚、大麻酚和内标物的定性离子对、定量
离子对和保留时间见表 2。毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的 MRM 色谱图参见附录 A。
表2 Δ9-四氢大麻酚、大麻二酚、大麻酚和内标物的定性离子对、定量离子对和保留时间
|
化合物
|
定性离子对
/(m/z)
|
定量离子对
/(m/z)
|
保留时间/
(min)
|
|
Δ9 -四氢大麻酚
|
315.2/193.2
|
315.2/193.2
|
2.69
|
|
315.2/259.1
|
|
大麻二酚
|
315.2/193.2
|
315.2/193.2
|
3.07
|
|
315.2/259.1
|
|
大麻酚
|
311.1/223.2
|
311.1/223.2
|
3.10
|
|
|
311.1/293.3
|
|
|
|
甲氧那明(内标)
|
180.2/148.8
|
180.2/148.8
|
7.76
|
6.1.2.2 进样
分别吸取案件样品、空白样品和添加样品提取液,按6.1.2.1条件进样分析。
6.1.2.3 记录
记录各样品中Δ9-四氢大麻酚、大麻二酚、大麻酚可疑色谱峰的保留时间和离子对丰度比。
6.1.2.4 定性判断依据
以保留时间、质谱特征碎片离子峰和离子对相对丰度比作为定性判断依据。
如果案件样品中出现Δ9-四氢大麻酚、大麻二酚、大麻酚的两对定性离子对的特征色谱峰,保留时
间与添加样品中相应标准物质的色谱峰保留时间比较,相对误差在±2.5 %内,且定性离子对丰度比与质
量分数相近添加样品的离子对丰度比之相对误差不超过表 3 规定的范围,则可认为样品中存在该种目标
物。
表3 相对离子对丰度比的最大允许相对误差(%)
|
相对离子对丰度比
|
>50
|
>20~50
|
>10~20
|
£10
|
|
允许的相对误差
|
±20
|
±25
|
±30
|
±50
|
6.2 定量分析
本技术规范采用内标法定量分析。
6.2.1 样品前处理
取毛发案件样品两份,按6.1.1.1操作。
另取空白毛发样品若干份,添加适量Δ9-四氢大麻酚、大麻二酚、大麻酚,制得系列质量分数或单
点质量分数的添加样品,与案件样品平行操作。方法学有效性验证数据参见附录B。
案件样品中Δ9-四氢大麻酚、大麻二酚、大麻酚的质量分数应在工作曲线的线性范围内。配制单点
质量分数的添加样品时,案件样品中Δ9-四氢大麻酚、大麻二酚、大麻酚质量分数需在添加样品质量分
数的±50 %内。
6.2.2 仪器检测
6.2.2.1 仪器参考条件同 6.1.2.1。
6.2.2.2 进样
分别将案件样品、系列质量分数的添加样品或单点质量分数添加样品,按6.1.2.1条件进样分析。
6.2.3 记录与计算
记录案件样品、系列质量分数的添加样品或单点质量分数添加样品中Δ9-四氢大麻酚、大麻二酚或
大麻酚及内标物的峰面积值,然后计算案件样品中Δ9-四氢大麻酚、大麻二酚或大麻酚含量。
6.2.3.1 内标-工作曲线法
在系列质量分数的添加样品中,以Δ9-四氢大麻酚、大麻二酚、大麻酚与内标物定量离子对的峰面
积比(Y)为纵坐标、Δ9-四氢大麻酚、大麻二酚、大麻酚质量分数(C)为横坐标进行线性回归,得线性方程。
根据案件样品中Δ9-四氢大麻酚、大麻二酚、大麻酚及内标物定量离子对的峰面积值,按公式(1)计
算出案件样品中Δ9-四氢大麻酚、大麻二酚或大麻酚的含量。

式中:
C ⎯⎯案件样品中目标物的质量分数,单位为纳克每毫克(ng/mg);
Y⎯⎯案件样品中目标物与内标物定量离子对的峰面积比;
a ⎯⎯线性方程的截距;
b ⎯⎯线性方程的斜率。
6.2.3.2 内标-单点校正法
根据案件样品和添加样品中Δ9-四氢大麻酚、大麻二酚、大麻酚及内标物定量离子对的峰面积值,
按公式(2)计算含量:

式中:
C ⎯⎯案件样品中目标物的质量分数,单位为纳克每毫克(ng/mg);
A⎯⎯案件样品中目标物与内标物的峰面积比值;
A′⎯⎯添加样品中目标物与内标物的峰面积比值;
c ⎯⎯添加样品中目标物的质量分数,单位为纳克每毫克(ng/mg)。
6.2.4 计算相对相差
案件样品按以上步骤平行测定两份,双样相对相差按公式(3)计算:
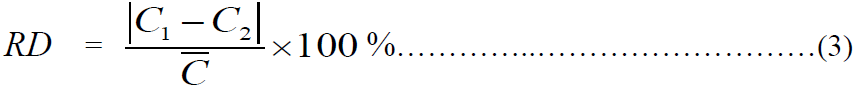
式中:
RD⎯⎯相对相差(%);
C1、C2⎯⎯两份案件样品平行定量测定的结果,单位为纳克每毫克(ng/mg);
C ⎯⎯两份案件样品平行定量测定结果的平均值(C1+ C2)/ 2,单位为纳克每毫克(ng/mg)。
7 分析结果评价
7.1 定性分析结果评价
7.1.1 阴性结果评价
如果案件样品中未检出Δ9-四氢大麻酚、大麻二酚、大麻酚,添加样品中检出Δ9-四氢大麻酚、大麻
二酚、大麻酚,则阴性结果可靠;如果添加样品中未检出Δ9-四氢大麻酚、大麻二酚、大麻酚,则阴性
结果不可靠,应按6.1重新提取检验。
7.1.2 阳性结果评价
如果案件样品中检出Δ9-四氢大麻酚、大麻二酚、大麻酚且空白样品无干扰,则阳性结果可靠;如
果空白样品亦呈阳性,则阳性结果不可靠,应按6.1重新提取检验。
7.2 定量分析结果评价
两份案件样品的相对相差不超过 20%时,结果按两份案件样品含量的平均值计算。否则需要重新
测定。
附 录 A
(资料性附录)
毛发中Δ9-四氢大麻酚、大麻二酚和大麻酚的 MRM 色谱图
A.1 空白头发MRM色谱图
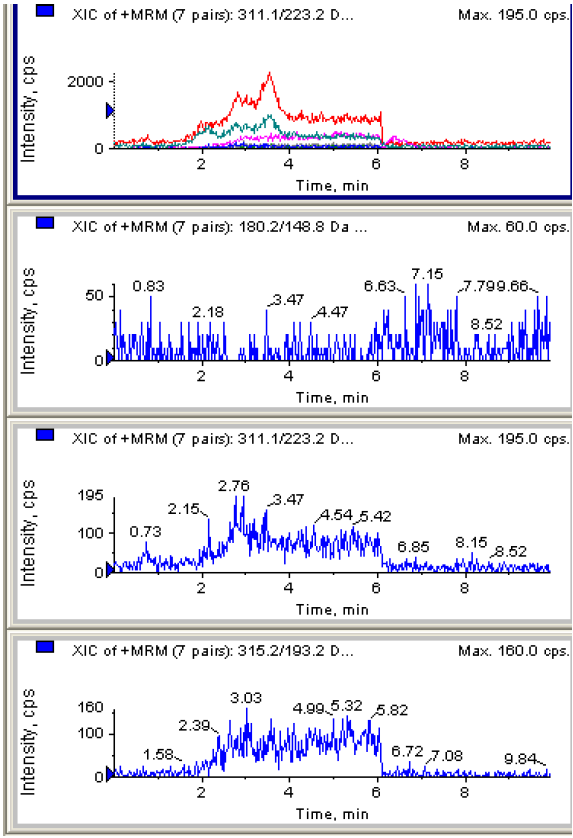
图A.1 空白头发 MRM 色谱图
A.2 头发添加样品的MRM色谱图
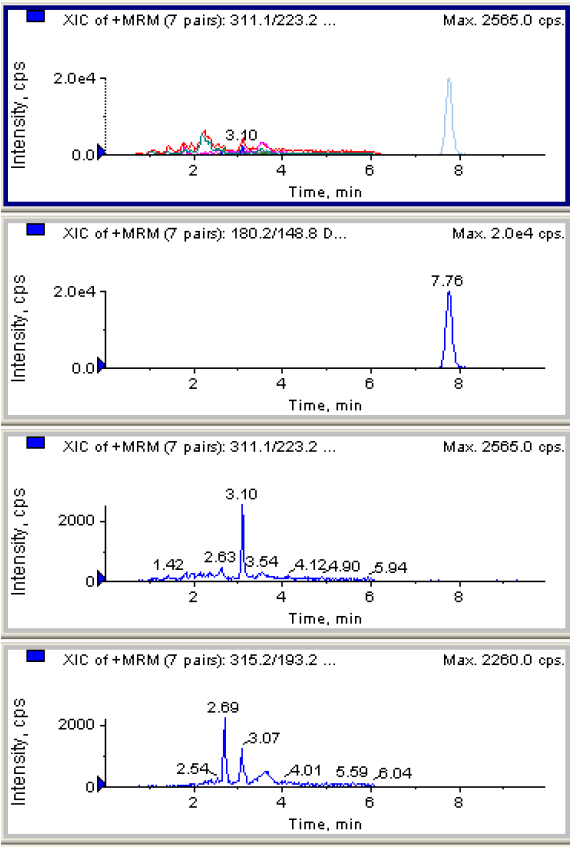
图A.2 空白头发中添加 0.05ng/mg 的Δ9-四氢大麻酚、大麻二酚和大麻酚 MRM 色谱图
附 录 B
(资料性附录)
方法学有效性验证数据
B.1 工作曲线、最低检出限(LOD)和定量下限(LOQ)
表B.1 工作曲线、最低检出限和定量下限
|
化合物
|
线性范围/(ng/mg)
|
线性方程
|
相关系数/ r
|
LOD/
(ng/mg)
|
LOQ/
(ng/mg)
|
|
Δ9 -四氢大麻酚
|
0.05~2.5
|
y=0.3716x-0.0057
|
0.9978
|
0.05
|
0.05
|
|
大麻二酚
|
0.05~2.5
|
y=0.1644x-0.0025
|
0.9963
|
0.05
|
0.05
|
|
大麻酚
|
0.05~2.5
|
y=0.8379x-0.0133
|
0.9956
|
0.05
|
0.05
|
B.2 方法准确度、精密度
表B.2 头发中Δ9 -四氢大麻酚、大麻二酚、大麻酚测定的精密度和准确度
|
化合物
|
添加样品含量/
(ng/mg)
|
准确度/ (%)
(n=24)
|
精密度/ RSD(%)
|
|
日内(n=6)
|
日间(n=24)
|
|
Δ9 -四氢大麻酚
|
0.05
|
108.1
|
4.4
|
19.5
|
|
0.4
|
107.7
|
6.0
|
15.2
|
|
2
|
102.8
|
5.6
|
14.6
|
|
大麻二酚
|
0.05
|
111.4
|
8.7
|
13.0
|
|
0.4
|
101.7
|
5.6
|
13.8
|
|
2
|
96.1
|
11.3
|
13.4
|
|
大麻酚
|
0.05
|
119.9
|
11.4
|
17.4
|
|
0.4
|
107.7
|
5.3
|
16.0
|
|
2
|
103.1
|
4.0
|
13.6
|



发表评论